Peutz–Jeghers syndrome, also known as hereditary intestinal polyposis syndrome, is an autosomal dominant genetic disease characterized by the development of benign hamartomatous polyps in the gastrointestinal tract and hyperpigmented macules on the lips and oral mucosa. Peutz–Jeghers syndrome has an incidence of approximately 1 in 25,000 to 300,000 births. Peutz-Jeghers syndrome (PJS) is named after two doctors who first studied and described it in 1921. It is an association of three very specific conditions in any one person.
Diagnosis
 The main criteria for clinical diagnosis are:
The main criteria for clinical diagnosis are:
- Family history
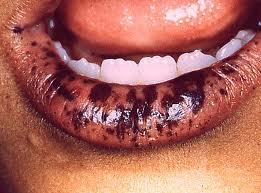
- Mucocutaneous lesions causing patches of hyperpigmentation in the mouth and on the hands and feet. The oral pigmentations are the first on the body to appear, and thus play an important part in early diagnosis. Intraorally, they are most frequently seen on the gingiva, hard palate and inside of the cheek. The mucosa of the lower lip is almost invariably involved as well.
- Hamartomatous polyps in the gastrointestinal tract. These are benign polyps with an extraordinarily low potential for malignancy.
Having 2 of the 3 listed clinical criteria indicates a positive diagnosis. The oral findings are consistent with other conditions, such as Addison’s disease and McCune-Albright syndrome, and these should be included in the differential diagnosis. 90-100% of patients with a clinical diagnosis of PJS have a mutation in the STK11/LKB1 gene. Molecular genetic testing for this mutation is available clinically.
Natural history
Most patients will develop flat, brownish spots (melanotic macules) on the skin, especially on the lips and oral mucosa, during the first year of life, and a patient’s first bowel obstruction due to intussusception usually occurs between the ages of six and 18 years. The cumulative lifetime cancer risk begins to rise in middle age. Cumulative risks by age 70 for all cancers, gastrointestinal (GI) cancers, and pancreatic cancer are 85%, 57%, and 11%, respectively.
Genetics
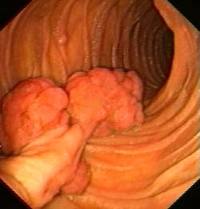
Gastrointestinal polyps
In 1998, a gene was found to be associated with the mutation. On chromosome 19, the gene known as STK11 (LKB1) is a possible tumor suppressor gene. It is inherited in an autosomal-dominant pattern which means that anyone who has PJS has a 50% chance of passing it onto their children, assuming that their spouse does not have the disease.
Limited evidence base
Peutz–Jeghers syndrome is rare and studies typically include only a small number of patients. Even in those few studies that do contain a large number of patients, the quality of the evidence is limited due to pooling patients from many centers, selection bias (only patients with health problems coming from treatment are included), and historical bias (the patients reported are from a time before advances in the diagnosis of treatment of Peutz–Jeghers syndrome were made). Probably due to this limited evidence base, cancer risk estimates for Peutz–Jeghers syndrome vary from study to study.
Presentation
The risks associated with this syndrome include a strong tendency of developing cancer in multiple sites. While the hamartomatous polyps themselves only have a small malignant potential (<3% – OHCM), patients with the syndrome have an increased risk of developing carcinomas of the pancreas, liver, lungs, breast, ovaries, uterus, testicles and other organs.
The average age of first diagnosis is 23, but the lesions can be identified at birth by an astute pediatrician. Prior to puberty, the mucocutaneous lesions can be found on the palms and soles. Often the first presentation is as a bowel obstruction from an intussusception which is a common cause of mortality; an intussusception is a telescoping of one loop of bowel into another segment.
Other symptoms are:
- Brownish or bluish-gray pigmented spots on the lips, gums, inner lining of the mouth, and skin
- Clubbed fingers or toes
- Cramping pain in the belly area
- Dark freckles on and around the lips of a newborn
- Blood in the stool that can be seen with the naked eye (occasionally)
- Vomiting
Cancer screening

Mammogram
Some suggestions for surveillance for cancer include the following:
- Small intestine with small bowel radiography every 2 years,
- Esophagogastroduodenoscopy and colonoscopy every 2 years,
- CT scan or  MRI of the pancreas yearly,
- Ultrasound of the pelvis (women) and testes (men) yearly,
- Mammography (women) from age 25 annually livelong, and
- Papanicolaou (Pap) test every year
Follow-up care should be supervised by a physician familiar with Peutz–Jeghers syndrome. Genetic consultation and counseling as well as urological and gynecological consultations are often needed.
Treatment
Surgery may be needed to remove polyps that cause long-term problems. Iron supplements help counteract blood loss.
Persons with this condition should be monitored by a health care provider and be checked periodically for cancerous polyp changes.
Outlook (Prognosis)
There may be a significant risk of these polyps becoming cancerous. Some studies link PJS and cancers of the gastrointestinal tract, lung, breast, uterus, and ovaries.