Progeria (also known as “Hutchinson–Gilford Progeria Syndrome“, “Hutchinson–Gilford syndrome“, and “Progeria syndrome“) is an extremely rare genetic condition wherein symptoms resembling aspects of aging are manifested at an early age. The word progeria comes from the Greek words “pro” (Ï€ÏÏŒ), meaning “before”, and “géras” (γῆÏας), meaning “old age”. The disorder has very low incidences and occurs in an estimated 1 per 8 million live births. Those born with progeria typically live to their mid teens and early twenties. It is a genetic condition that occurs as a new mutation (de novo), and is rarely inherited. Although the term progeria applies strictly speaking to all diseases characterized by premature aging symptoms, and is often used as such, it is often applied specifically in reference to Hutchinson-Gilford Progeria Syndrome (HGPS).
Scientists are particularly interested in progeria because it might reveal clues about the normal process of aging. Progeria was first described in 1886 by Jonathan Hutchinson. It was also described independently in 1897 by Hastings Gilford. The condition was later named Hutchinson-Gilford Progeria Syndrome (HGPS).
Signs and symptoms
 Children with progeria usually develop the first symptoms during infancy. The earliest symptoms include failure to thrive and a localized scleroderma-like skin condition. As a child ages past infancy, additional conditions usually become apparent around 18–24 months. Limited growth, full-body alopecia, and a distinctive appearance (small face and jaw, pinched nose) are all characteristics of progeria. Signs and symptoms of this progressive disease tend to get worse as the child ages. Later, the condition causes wrinkled skin, atherosclerosis, kidney failure, loss of eyesight, hair loss, and cardiovascular problems. Scleroderma, a hardening and tightening of the skin on trunk and extremities of the body, is prevalent. People diagnosed with this disorder usually have small, fragile bodies, like those of elderly people. The face is usually wrinkled, with a larger head in relation to the body, a narrow face and a beak nose. Prominent scalp veins are noticeable (made more obvious by hair loss), as well as prominent eyes. Musculoskeletal degeneration causes loss of body fat and muscle, stiff joints, hip dislocations, and other symptoms generally absent in the non-elderly population. Individuals usually retain normal mental and motor development.
Children with progeria usually develop the first symptoms during infancy. The earliest symptoms include failure to thrive and a localized scleroderma-like skin condition. As a child ages past infancy, additional conditions usually become apparent around 18–24 months. Limited growth, full-body alopecia, and a distinctive appearance (small face and jaw, pinched nose) are all characteristics of progeria. Signs and symptoms of this progressive disease tend to get worse as the child ages. Later, the condition causes wrinkled skin, atherosclerosis, kidney failure, loss of eyesight, hair loss, and cardiovascular problems. Scleroderma, a hardening and tightening of the skin on trunk and extremities of the body, is prevalent. People diagnosed with this disorder usually have small, fragile bodies, like those of elderly people. The face is usually wrinkled, with a larger head in relation to the body, a narrow face and a beak nose. Prominent scalp veins are noticeable (made more obvious by hair loss), as well as prominent eyes. Musculoskeletal degeneration causes loss of body fat and muscle, stiff joints, hip dislocations, and other symptoms generally absent in the non-elderly population. Individuals usually retain normal mental and motor development.
Cause
|
Steps in normal cell |
Steps in cell with progeria |
| The gene LMNA encodes a protein called prelamin A. | The gene LMNA encodes a protein called prelamin A. |
| Prelamin A has a farnesyl group attached to its end. | Prelamin A has a farnesyl group attached to its end. |
| Farnesyl group is removed from prelamin A. | Farnesyl group remains attached to prelamin A. |
| Normal form is called prelamin A. | Abnormal form of prelamin A is called progerin. |
| Prelamin A is not anchored to the nuclear rim. | Progerin is anchored to the nuclear rim. |
| Normal state of the nucleus. | Abnormally shaped nucleus. |
 In normal conditions, the LMNA gene encodes a protein called prelamin A, which leads to the lamin A form. There is a farnesyl group attached to the carboxyl-terminus of its structure. Farnesyl molecule allows prelamin A to be anchored to nuclear rim, and then it is removed. Before the late 20th century, research on progeria yielded very little information about the syndrome. In 2003, the cause of progeria was discovered to be a point mutation in position 1824 of the LMNA gene, replacing cytosine with thymine, creating a truncated form, progerin, of the prelamin A protein whose further processing is abnormal. Normal lamin A is a major structural protein of the nuclear lamina of the nucleus (along with lamin C);when LMNA is mutated, farnesyl group can not be removed and progerin does not integrate into this structure and accumulates, disrupting it. This is when HGPS appears. Since the nuclear lamina participates in organizing chromatin and supporting the nuclear envelope; this disrupts the human cell nucleus as a whole, limiting the ability of the cell to divide. Then, the appearance of affected nuclei is like bubbles or a cluster of grapes. Progerin may also play a role in normal human aging, since its production is activated in senescent wild type cells.
In normal conditions, the LMNA gene encodes a protein called prelamin A, which leads to the lamin A form. There is a farnesyl group attached to the carboxyl-terminus of its structure. Farnesyl molecule allows prelamin A to be anchored to nuclear rim, and then it is removed. Before the late 20th century, research on progeria yielded very little information about the syndrome. In 2003, the cause of progeria was discovered to be a point mutation in position 1824 of the LMNA gene, replacing cytosine with thymine, creating a truncated form, progerin, of the prelamin A protein whose further processing is abnormal. Normal lamin A is a major structural protein of the nuclear lamina of the nucleus (along with lamin C);when LMNA is mutated, farnesyl group can not be removed and progerin does not integrate into this structure and accumulates, disrupting it. This is when HGPS appears. Since the nuclear lamina participates in organizing chromatin and supporting the nuclear envelope; this disrupts the human cell nucleus as a whole, limiting the ability of the cell to divide. Then, the appearance of affected nuclei is like bubbles or a cluster of grapes. Progerin may also play a role in normal human aging, since its production is activated in senescent wild type cells.
Unlike “accelerated aging diseases” (such as Werner’s syndrome, Cockayne’s syndrome, or xeroderma pigmentosum), progeria is not caused by defective DNA repair. Because these diseases display what are considered different aspects of aging, but never every aspect, they are often called “segmental progerias”.
Diagnosis
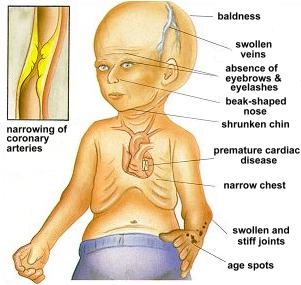 Diagnosis is suspected according to signs and symptoms, such as skin changes, abnormal growth, and loss of hair. A genetic test for LMNA mutations can confirm the diagnosis of progeria.
Diagnosis is suspected according to signs and symptoms, such as skin changes, abnormal growth, and loss of hair. A genetic test for LMNA mutations can confirm the diagnosis of progeria.
Treatment
No treatments have been proven effective. Most treatment focuses on reducing complications (such as cardiovascular disease) with heart bypass surgery or low-dose aspirin. Children may also benefit from a high-calorie diet.
Growth hormone treatment has been attempted. The use of morpholinos has also been attempted in order to reduce progerin production. Antisense morpholino oligonucleotides specifically directed against the mutated exon 11–exon 12 junction in the mutated premRNAs were used.